
Metaphyseal Chondrodysplasia Schmid type - widening the phenotype: Significant variable expression in two familial cases with heterozygous COL10A1 variants
AH Sabir1*, S Mahesh2, RT Freeman3, N Kiely3, A Mageuri4, D Lim5, T Cole5
1Clinical Genetics Registrar, Birmingham Women’s and Children’s Hospital NHS Trust and Birmingham Health Partners, UK
2Foundation Doctor, Royal Wolverhampton Hospital NHS Trust, UK
3Paediatric Orthopaedic Consultant. Robert Jones & Agnes Hunt Orthopaedic Hospital, Oswestry, UK
4Clinical Genetic Scientist, Department of Clinical Genetics, Amsterdam UMC, Vrije Universiteit Amsterdam, The Netherlands
5Consultant Clinical Geneticist. Birmingham Women’s and Children’s Hospital NHS Trust and Birmingham Health Partners, UK
Abstract
Metaphyseal Chondrodysplasia Schmid Type (SMCD) is a rare dominant form of skeletal dysplasia, caused by heterozygous mutations in the COL10A1 (Collagen Type X alpha 1) gene which codes for α1 chains in type X collagen, and typically results in disproportionate short stature. We report two families with the clinical and radiological diagnosis of SMCD seen in our unit over 4 years. In the first family (A), the proband had a possible pathogenic COL10A1 variant (c.133C>T, p.Pro45Ser). However, his father had the same variant without any obvious signs of SMCD, causing uncertainty whether this mutation was pathogenic. In the second family (B), the proband and her affected father had the same variant. Almost at the same time, a large consanguineous family was reported to have segregation of the variant with SMCD phenotypes in that the homozygosity was related to severer phenotypes and the heterozygosity to milder phenotypes. Hence, we were convinced that the variant was pathogenic. The pathogenic variant (p.Pro45Ser) causes a wide phenotypic spectrum of SMCD and even subclinical features. In this report, we highlight attenuated phenotypes of SMCD, and also discuss the latest therapeutic advances in SMCD and its mechanism of action.
Introduction
Metaphyseal chondrodysplasia encompasses a heterogeneous group of rare skeletal dysplasias. Metaphyseal chondrodysplasia Schmid type (SMCD) is the most common type of the group (others include Jansen and McCusick MCD). SMCD manifests with disproportionate short stature with rhizomelic shortening of the limbs and waddling gait in early childhood1. The classical radiological features include widened growth plates, metaphyseal irregularities, and metaphyseal flaring of the long bones, particularly at the knees and hips. Coxa vara is a common feature. SMCD is an autosomal dominant condition caused by heterozygous mutations of COL10A1 which codes for type X collagen. Increasing availability of molecular testing for genetic disorders has increased the likelihood of detecting variants of unknown pathogenic significance. This report on two families with SMCD due to a pathogenic variant (p.Pro45Ser) highlights the attenuated phenotypes and intrafamilial phenotypic variations in these families, causing uncertainty as to whether the genetic variant was of pathological significance.
Methods
This article is a retrospective report of two families referred to the West Midlands Regional Clinical Genetics Unit, between 2014 to 2018. In both families, the probands showed progressive genu valgum presumably due to a genetic bone disease, and the family histories were notable. The data of the medical history, clinical examination, and sequential growth parameters were retrieved from the clinical records. Additional diagnostic work-up included serum biochemical testing (including serum alkaline phosphatase, calcium, phosphate and vitamin D levels) and radiographic imaging, including the radiographs of the lower limbs and pelvic in both probands, those of the knees and pelvis in the proband’s father in the first family (Family A) and those of the left knee and left hip in the proband’s father in the second family (Family B). A single gene (COL10A1) DNA Sanger sequencing for the probands and follow-up targeted sequencing for the parents was performed. DNA testing was conducted at the Genome Diagnostic lab at the VU University Medical Centre, Amsterdam.
Results
Family A
A 7-year-old boy (P1), the second child of three, born to unrelated Caucasian parents presented with unresolved genu valgum and a waddling gait aged 4 years. He was born at 37 weeks of gestation via normal vaginal delivery. Prenatal imaging had revealed gastroschisis and an umbilical hernia, which were surgically repaired after birth. He had a family history of early-onset hip replacement in his maternal grandmother and great aunt (both at age 52 years), as shown in the pedigree (figure 1). Growth parameters aged 7 were as follows: height 122 cm (50th centile) and OFC (occipto-frontal circumference) 55 cm (97th centile). Weight at age 5 years was 22 kg (>75th centile). His facial features were normal. Psychomotor developmental was normal. He had marked bilateral, asymmetric genu valgum. Both hips abducted to only ~30 degrees. The genu valgum was corrected using a surgical technique of guided growth using an eight plate and screws (image 1.1).
Investigations
Biochemical testing, including ALP (alkaline phosphatase), phosphate and Vitamin D, was normal. Radiographs are described in Image 1.1a-b.
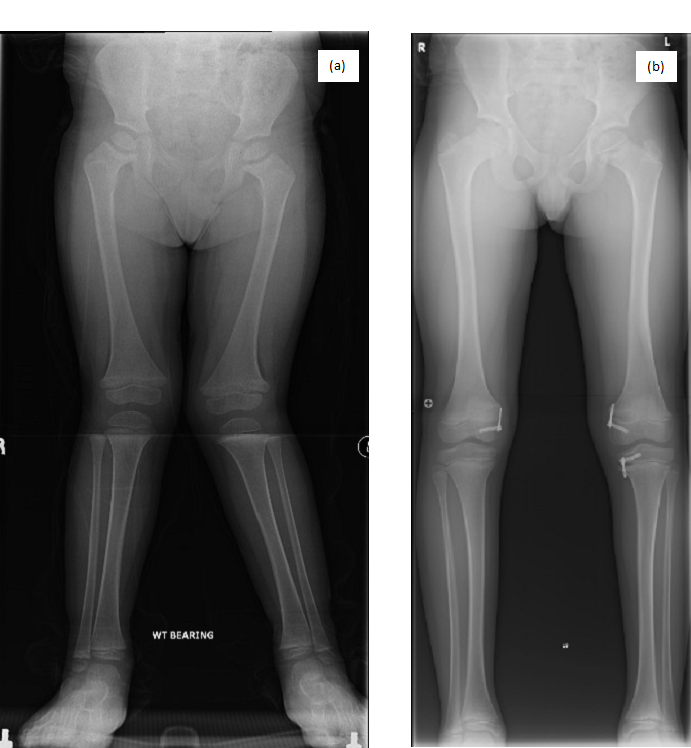
Image 1.1 (a and b): Lower limb AP radiographs of P1. (a) Radiograph of P1 before surgery (aged 4 years) showing bilateral genu valgum, coxa vara, metaphyseal widening, cupping, and irregularities of the proximal and distal femora and metaphyseal widening of the proximal tibiae. (b) Radiograph of P1 after surgery (aged 10 years) showing correction of genu valgum.
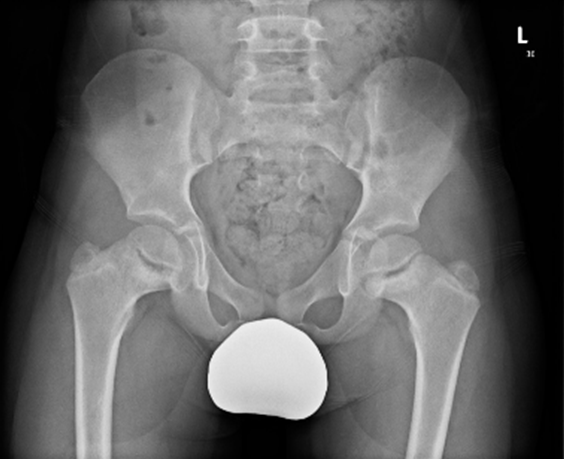
Image 1.2 Pelvic radiograph of P1 after surgery (aged 8 years) showing metaphyseal abnormalities of the proximal femora (ragged and cupped proximal metaphyses) and bilateral coxa vara.
The clinical presentation and radiographic findings suggested SMCD. Sanger sequencing for all exons and surrounding splice sites of COL10A1 revealed a novel heterozygous missense variant c.133C>T (p.Pro45Ser, substitution of serine for proline in a non-collageneous NC2 domain of COL10A1). The proline is highly conserved across species. However, most of the previously described pathogenic COL10A1 variants have been reported in the other non-collageneous NC1 domain. Based on these considerations, the variant was classified as a variant of uncertain clinical significance (VUS). A DNA analysis for the parents indicated that it was paternally transmitted. However, his father was deemed to be asymptomatic with a normal stature (178cm) and previous radiographic examination was reported as normal (image 1.2a-b). Therefore, this missense variant was considered unlikely to be pathogenic.
.png)
Image 1.2 (a and b): Radiographs from P1’s father.
(a) Knee radiograph interpreted as ‘normal’ (aged 39).
(b) Pelvic radiograph was reported as ‘normal’ (aged 36) but review with calculations (Image 1.2c-d) showed ‘cervical’ coxa vara (a subtle sign of SCMD) and mild degenerative changes.
(c and d) calculation of ‘cervical’ coxa vara with abnormal medial proximal femoral angle (MPFA) of 68o, left and 70o, right (normal 80-90 o) and reduced femoral head/neck ratios of 0.696, left and 0.666, right (normal 0.27 ±0.03)2,3.
However, retrospective radiological assessment for the hip (image 1.2 b, c, d) ascertained subtle ‘cervical’ coxa vara and femoral neck shortening, suggestive of metaphyseal dysplasia.
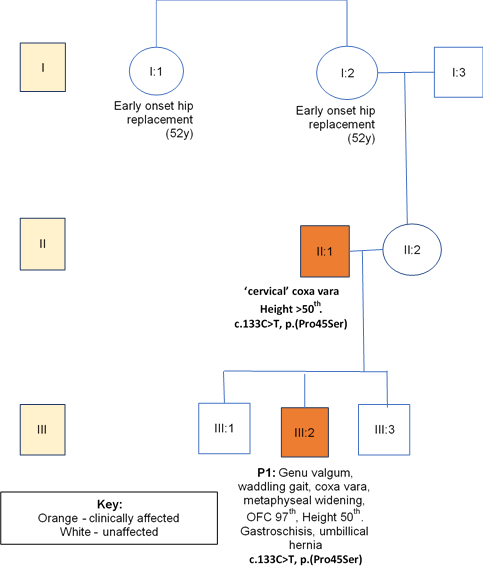
Figure 1: Family A with co-segregation and phenotypic data.
Family B
A 4-year-old female (P2), the second child of three, born to unrelated Caucasian parents, presented with short stature and genu valgum. She was born at 38 weeks following an uneventful pregnancy and normal vaginal delivery. At 3 years 9 months, height was 96cm (9th centile). She showed normal facial features and normal head size. She showed mild upper-limb rhizomelia and symmetrical progressive genu valgum. She underwent a guided growth surgery at her knees, as shown in image 2.1. Her father and paternal aunt required multiple surgeries for bowed legs and they were 168cm and 150cm tall, respectively, as detailed in the pedigree (figure 2). Her father showed a normal knee joint after genu valgum correction surgery (image 2.2a), but demonstrated shortening of the femoral necks and mild degenerative changes of the hip joint (image 2.2b).
Investigations
Biochemical testing including ALP, phosphate and Vitamin D was normal. Radiographs described in image 2.1.
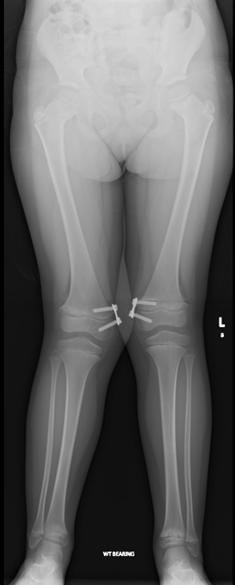
Image 2.1 Lower limb radiograph of P2 after surgery of a guided growth technique at the knees to correct genu valgum (aged 6 years) showing proximal femoral metaphyseal irregularities with femoral neck broadening and distal femoral metaphyseal widening.
.png)
Image 2.2 (a and b) Radiographs of the father of P2. (a) Radiograph of the left knee (aged 46) following osteotomy and plate fixation to correct genu valgum showing mild degenerative changes. (b) Radiograph of the left hip showing reduced proximal femoral epiphyseal height with a short femoral neck (Aged 46).
The clinical and radiological features suggested SMCD. COL10A1 testing identified the same variant (paternally inherited) as in family A.
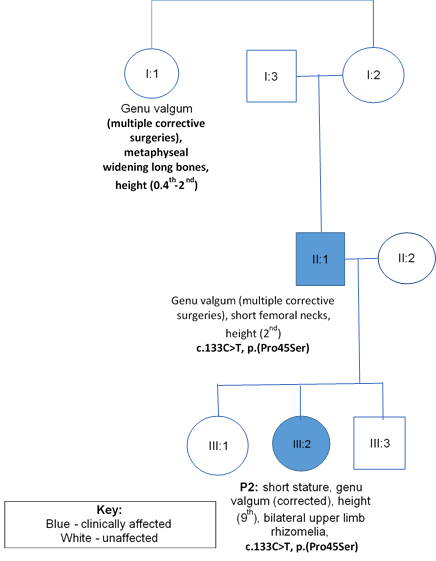
Figure 2: Family B with co-segregation and phenotypic data
Discussion
SMCD is caused by mutations in COL10A1 which codes for α1-chains in type X collagen (structure described in figure 3). It is a network forming collagen, largely expressed in hypertrophic chondrocytes, mainly hypertrophic zones of growth plates and articular-cartilage of long bones4. Thus, abnormal type X collagen leads to growth plate defects and possibly defective articular cartilage5. To date, 47 pathogenic COL10A1 variants have been reported, 45 within the globular domain (NC1)6,7,8,9.
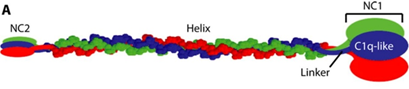
Figure 3: Image taken from Horton et al [under creative commons open access license]. Type X Collagen molecule with central triple helix homotrimer (three α1 chains) flanked by an amino-terminal domain (NC2) and a globular domain (NC1).
http://stm.sciencemag.org/content/9/419/eaan4669
In this report, both probands with SMCD had a paternally inherited variant, c.133C>T (p.Pro45Ser), substituting a serine residue for a highly conserved proline residue in the NC2 domain. In general, it is not straightforward to make a radiological diagnosis for adults with genetic bone diseases. Nevertheless, meticulous radiological assessment ascertained that the fathers were also affected. Additionally, Ain N et al, recently reported a large consanguineous family with p.Pro45Ser8, in which the homozygous state caused a severe form of chondrodysplasia, whilst heterozygous carriers had a mild phenotype of short stature. It is now clear that c.133C>T (p. Pro45Ser) is a pathogenic variant. However, the phenotype of the father in Family A was so mild on clinical and radiological grounds that c.133C>T (p.Pro45Ser) was initially regarded as unlikely to be a pathogenic variant. Height of adult patients with SMCD is generally less than 3.5SD of the mean, although a number of patients with normal height have been reported9. In this series both probands were not particularly short. Our report illustrated the wider clinical variability associated with the (p.Pro45Ser) variant and also highlight the large phenotypic spectrum of SMCD, which should be taken in consideration in the interpretation of hitherto uncharacterised molecular findings in COL10A1.
It was intriguing to see premature degenerative disease in older family members of Family A and to a lesser degree in the father of Family B. It is a matter of debate whether this feature should be part of the SMCD phenotype. Interestingly the clinical synopsis of SMCD in the OMIM database specifically mentions ‘no osteoarthritic symptoms’ as a clinical feature. This report stresses the need for careful phenotypic analysis to confirm diagnosis. This is increasingly important with the emergence of potential treatments.
Treatments for SMCD are limited to supportive physiotherapy, occupational therapy and surgical correction for lower limb deformities. More recently there have been drug trials looking at the efficacy of Carbamazepine treatment. Carbamazepine (CBZ), is a well-known anti-epileptic drug, is currently in trials for SMCD due to its ability to reduce ER (endoplasmic reticulum) stress (as discussed in Table 1). Mutations in COL10A1 leads to retention of misfolded collagen X within the ER of hypertrophic chondrocytes. Increased ER stress then leads to the unfolded protein response and ultimately disruption of the normal growth plate and bone growth10. CBZ efficacy has been demonstrated in SMCD mouse models showing a reduction in severity of phenotypic features10. More information can be found on the trial website10.
Table 1: Emerging Therapies in SMCD; MCDS-Therapy (international investigator led) trial10
|
Drug / Phase |
Molecular abnormality |
Mechanism |
Recruitment |
|
Carbamazepine (phase 1-2) |
Misfolded collagen X retained within hypertrophic cartilage ER leads to ER stress and unfolded protein response à growth plate disruption à abnormal bone growth. |
CBZ reduces ER stress by stimulating intracellular degradation through autophagy or proteasomal degradation. |
Recruitment opened in April 2019 for ambulant children with SMCD up to age 11 years (before growth plates fuse). |
Acknowledgements
No funding.
Conflict of Interest
No conflicts of interests to declare.
References
- Al Kaissi A, Ghachem MB, Nabil NM, et al. Schmid’s Type of Metaphyseal Chondrodysplasia: Diagnosis and Management. Orthop Surg [Internet]. 2018; 10(3): 241–6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30027601
- Paley D. Principles of Deformity Correction [Internet]. Berlin, Heidelberg: Springer Berlin Heidelberg; 2002 [cited 2019 Nov 6]. Available from: http://link.springer.com/10.1007/978-3-642-59373-4
- Belangero PS, Bastos TA, Linhares GK, et al. Comparison of the Femoral Head Height/Neck Length Ratio between the Unaffected Hip of Patients with a Unilateral Slipped Femoral Head and The Hips of Individuals without a Slipped Femoral Head. Rev Bras Ortop (English Ed [Internet]. 2011 Jan [cited 2019 Nov 6]; 46(1): 57–63. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27026987
- Gudmann NS, Karsdal MA. Biochemistry of Collagens, Laminins and Elastin. In Karsdal MA editor Academic Press. 2016; 73–6. Available from: http://www.sciencedirect.com/science/article/pii/B9780128098479000106
- Bateman JF, Wilson R, Freddi S, et a;. Mutations of COL10A1 in Schmid metaphyseal chondrodysplasia. Hum Mutat [Internet]. 2005 Jun 1 [cited 2019 Mar 5]; 25(6): 525–34. Available from: http://doi.wiley.com/10.1002/humu.20183
- Institute of Medical Genetics Cardiff. The Human Gene Mutation Database [Internet]. [cited 2019 Mar 17]. Available from: http://www.hgmd.cf.ac.uk/ac/gene.php?gene=COL10A1
- Ikegawa S, Nakamura K, Nagano A, et al. Mutations in the N-terminal globular domain of the type X collagen gene (COL10A1) in patients with Schmid metaphyseal chondrodysplasia. Hum Mutat [Internet]. 1997 [cited 2019 Mar 17]; 9(2): 131–5. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9067753
- Ain N ul, Makitie O, Naz S. Autosomal recessive chondrodysplasia with severe short stature caused by a biallelic COL10A1 variant . J Med Genet. 2017; jmedgenet-2017-104885.
- Richmond CM, Savarirayan R. Schmid Metaphyseal Chondrodysplasia [Internet]. GeneReviews®. University of Washington, Seattle; 2019 [cited 2019 Nov 8]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/31633898
- MCDS therapy - Towards treatment of this rare bone disease [Internet]. [cited 2019 Jun 7]. Available from: https://mcds-therapy.eu/